News Story
Modeling Cellular Membranes
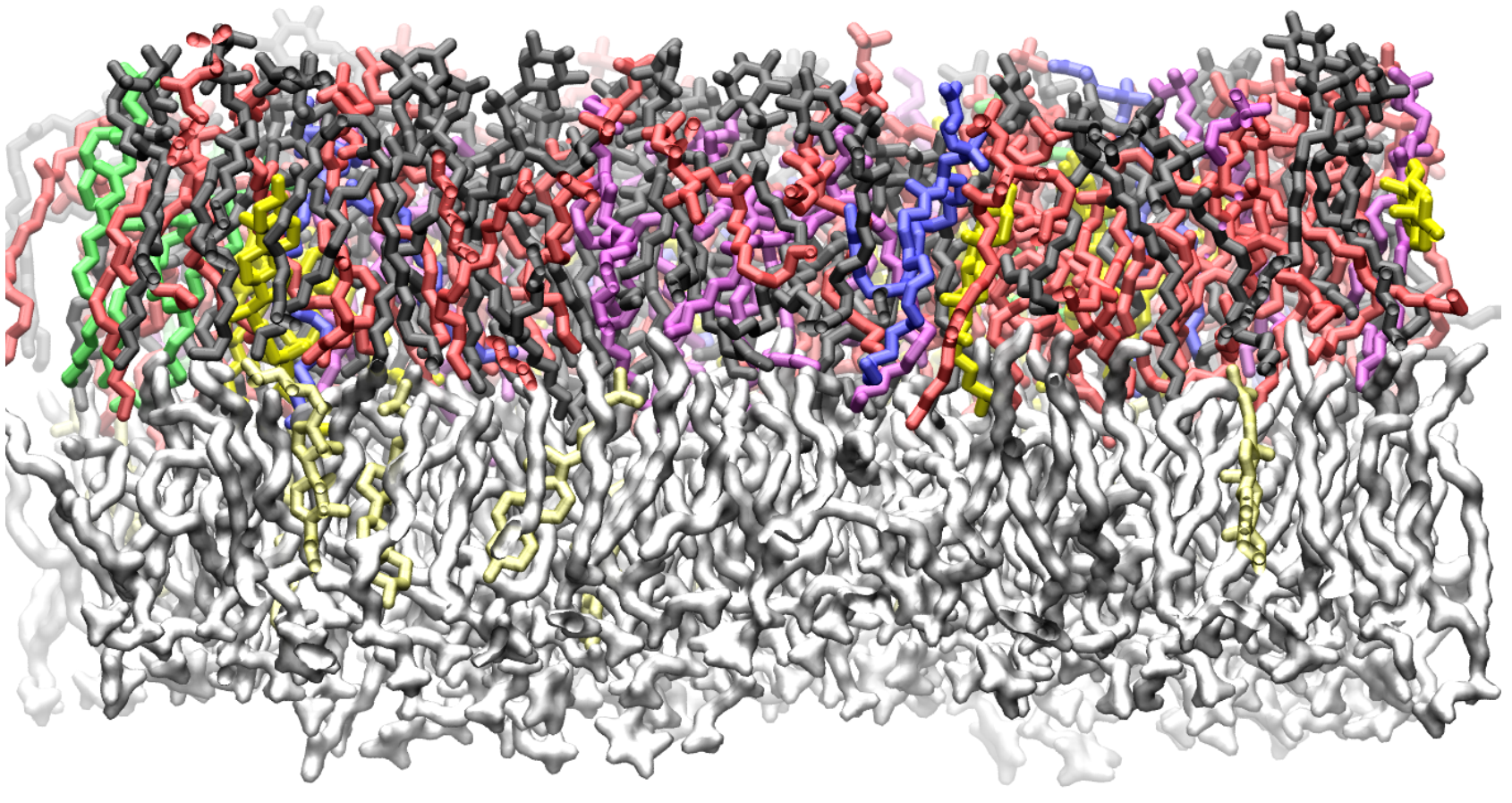
Computer simulations of membranes, composed primarily of phospholipids, are a popular tool for investigating structural, dynamic and thermodynamic properties of membranes and for probing lipid-protein interactions.
In an article recently published in Chemical Reviews, a research team at the University of Maryland (UMD) led by Jeffery Klauda, an associate professor in the Department of Chemical and Biomolecular Engineering (ChBE), evaluates the development and current states of commonly used lipid force-fields – the potential energy function paired together with various parameters – including parameterization goals and methods, and types of lipids each force-field can currently model.
Current uses of lipid force-fields range from theoretical studies of membrane phase behavior to applications in drug development; thus, the continued development of these force-fields is critical to the accuracy of computer simulations to ensure every atom is accounted for. UMD Ph.D. student, Alison Leonard, served as the paper’s fist author.
“We discussed the time and length scales accessible to force fields with various levels of atomic detail,” said Leonard. “For example, simulations with interaction sites for each atom frequently reach the microsecond timescale and can include proteins and ligands in a diverse lipid environment with water and ions.”
Phospholipids – such as lecithin, commonly found in soybean and egg products – are the primary components of biological membranes, having both a hydrophilic head-group and hydrophobic tails (picture a tadpole with two tails). A membrane is formed when two sheets of phospholipids align tail-to-tail, so that the head-groups face outward. Water-based solutions favor the head groups, but there is a large energy barrier for polar groups, such as water or ions, to cross the membrane.
“In this way, membranes separate solutions with different properties and maintain concentration gradients essential to cell viability,” Leonard explained. “Processes such as protein folding and ligand binding, as well as the flow of various species through membrane pores, occur in the context of a membrane and are influenced by composition and phase behavior.”
“Understanding the molecular structure of such membranes deepens our understanding of one of life's most fundamental aspects, and can also guide therapeutic development,” said co-author Eric Wang, a bioengineering student at UMD. “For example, our simulations could be used to study potential drugs that interact with, or pass through membranes, a common feature of dermal patches. In the future, faster computers and more accurate force-fields may even allow us to simulate structures on the cellular level with atomic resolution.”
With the rapid improvement of technology, thus, computational methods, simulating complex membranes will likely move from state-of-the-art to being common approaches to the study of cell membranes.
“In the next decade or two, it will likely be possible to simulate a simple whole cell” said Leonard. “This is a thrilling time to be part of a community working on aiding in the understanding of these complex assemblies, and their implications to various diseases.”
For additional information:
Leoard, A., et al (2019). Developing and Testing of Lipid Force Fields with Applications to Modeling Cellular Membranes (Chem. Rev. 2019, 119, 6227 – 6269). DOI: 10.1021/acs.chemrev.8b00384.
Published June 24, 2019
Related Stories
Stories / November 5, 2019
'Leapfrog' Model Offers New Insight into Transdermal Drug...
Stories / June 25, 2026
Grand Challenges 2.0: Engineers Partner to Unlock Solutions

Stories / May 13, 2026
Capstone Design Expo 2026: Students Solve for Maryland & Beyond

Stories / September 15, 2025
Sprayable Hydrogel Shows Promise for Overcoming Drug Delivery...

Stories / April 25, 2025
76 Undergrads Recognized at Annual Honors & Awards Celebration

Stories / May 8, 2024
MARC Program Now Accepting Applications
Stories / December 17, 2021
Nine Maryland Engineers Recognized as Being "One in 1,000"

Stories / November 20, 2019
Refocusing In-situ Electron Microscopy
Stories / November 20, 2019
MEI² Scientists Named Highly Cited Researchers in 2019

Stories / October 26, 2019
What’s Next for Next-Gen Batteries?